Сравнительный анализ метилирования сайтов RCGY в геномах клеточных линий L-68, Raji и U-937
Абдурашитов М.А.1,*, Томилов В.Н.1, Гончар Д.А.1, Снежкина А.В.2, Краснов Г.С.2, Кудрявцева А.В. 2,3, Дегтярев С.Х.1
1 СибЭнзайм, Новосибирск
2 Институт молекулярной биологии им. В.А. Энгельгардта, Москва
3 ФГБУ «Национальный медицинский исследовательский центр радиологии» Министерства здравоохранения РФ
* Автор для переписки: Абдурашитов М.А., НПО «СибЭнзим», Новосибирск 630117, ул. Ак. Тимакова 2/12; Тел. 3833334991, Факс 3833336853; E-mail: abd@sibenzyme.ru
В настоящей работе представлены результаты картирования метилированных сайтов RCGY в геномах двух малигнантных клеточных линий и контрольной клеточной линии фибробластов легких. Проведенный анализ выявил существенные отличия по метилированию генов, повторов, CpG-островков между тремя геномами. Обнаружены различия по степени метилирования различных групп повторов, которые, возможно, могут быть использованы в диагностических целях при условии разработки достаточно чувствительных методов анализа. Создана база данных, содержащая результаты картирования метилированных сайтов в геномах. Полученные данные по метилированию регуляторных участков различных генов, сопоставленные с ранее опубликованными данными, могут быть использованы для поиска маркеров метилирования для диагностики онкологических и некоторых других заболеваний. Таким образом, разработанный простой и нетрудоемкий метод картирования метилированных сайтов R(5mC)GY в геномах может быть использован на практике.
Цитирование: Абдурашитов М.А., Томилов В.Н., Гончар Д.А., Снежкина А.В., Краснов Г.С., Кудрявцева А.В., Дегтярев С.Х. (2019) Сравнительный анализ метилирования сайтов RCGY в геномах клеточных линий L-68, Raji и U-937 Эпигененическая ДНК диагностика, том 2019(1), DOI: 10.26213/SE.2019.76.40116
ВВЕДЕНИЕ
В настоящее время значительно вырос интерес к анализу метилирования геномной ДНК, что вызвано важной ролью этой эпигенетической модификации для нормального функционирования клеток. Особенное внимание при этом уделяется аберрантному метилированию различных участков ДНК, возникающим при малигнизации клеток, поскольку такие нарушения могут быть использованы для диагностики онкологических заболеваний [1-3]. Однако, накоплению данных по метилированию геномной ДНК препятствует ряд трудностей, связанных с недостатками используемых методов, а также с трудоемкостью и дороговизной такого рода исследований [4-5].
Ранее нами был предложен простой метод определения положения метилированных сайтов R(5mC)GY в геноме человека, основанный на расщеплении ДНК метилзависимой ДНК-эндонуклеазой GlaI и секвенировании образуемых фрагментов генома на NGS-приборе. Этот метод был использован для установления значительного числа метилированных сайтов в геноме клеточной линии Raji с помощью секвенирования на приборе Illumina MiSeq. Анализ результатов показал хорошее соответствие с данными, полученными при локус-специфическом определении статуса метилирования ряда генов-онкосупрессоров путем GlaI-ПЦР-анализа [6].
Целью настоящей работы являлось проведение более масштабного секвенирования GlaI-фрагментов геномов малигнантных клеточных линий Raji и U-937, а также немалигнантной клеточной линии L-68 для выявления различий в метилирования сайтов RCGY.
МАТЕРИАЛЫ И МЕТОДЫ
Препараты геномной ДНК клеточных линий L68, Raji и U937, а также метилзависимая сайт-специфическая ДНК-эндонуклеазой GlaI производства НПО «СибЭнзим» (Новосибирск). Гидролиз ДНК ферментом GlaI проводили в услових, рекомендованных производителем. Продукты гидролиза разделяли электрофорезом в 1,4% агарозном геле в трис-ацетатном буфере. GlaI-фрагменты длиной 140-400 п.н. вырезались из геля и очищались с помощью набора “Cyt 202” («Цитокин», Санкт-Петербург). Подготовку фрагментов ДНК к секвенированию осуществляли согласно протоколу производителя инструмента Genome Analyzer IIx (Illumina, США). В процессе секвенирования определялись нуклеотидные последовательности обоих концов фрагментов по 75 п.н.
Полученные риды были отфильтрованы с целью удаления последовательностей с низким качеством чтения, а также последовательностей, не содержащих динуклеотиды GY на 5′-концах, и, следовательно, не являющихся продуктами гидролиза ферментом GlaI.
Нуклеотидная последовательность референсного генома человека (сборка GRCh38.p12), координаты генов (Gencode release 23 [7]), кодирующих участков генома (CDS), CpG-островков и ДНК-повторов были получены с сайта » UCSC Genome Bioinformatics» [8]. Картирование ридов на референсный геном и визуализацию результатов проводили с помощью программного обеспечения «CLC Genomics Workbench 8.5» (Qiagen Aarhus A/S, Aarhus, Дания).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
В результате секвенирования концов GlaI-фрагментов длиной 140-400 п.н.было получено приблизительно по 100 млн ридов для каждого из трех геномов. Результаты фильтрации и картирования полученных ридов приведены в таблице 1.
Таблица 1
Результаты фильтрации и картирования ридов на референсный геном.| Геном | Всего ридов | После фильтрации | Картировано на референсный геном | Покрытая часть генома | Средняя глубина покрытия (без учета непокрытых областей) |
|---|---|---|---|---|---|
| L-68 | 100 362 080 | 79 626 274 | 72 144 184 | 12% | 14,7 |
| Raji | 94 526 060 | 70 252 160 | 61 364 793 | 10% | 14,9 |
| U-937 | 103 712 902 | 66 477 816 | 59 179 479 | 10% | 14,24 |
Для оценки покрытия различных участков генома проводили сравнение числа надежно покрытых участков (p-значение <0,005) на участках хромосом по 1 млн п.н. с количествами генов, кодирующих областей (CDS), CpG-островков, потенциальных сайтов узнавания GlaI (RCGY) и ДНК-повторов. Пример сравнения для хромосомы 1 представлен на рисунке 1.
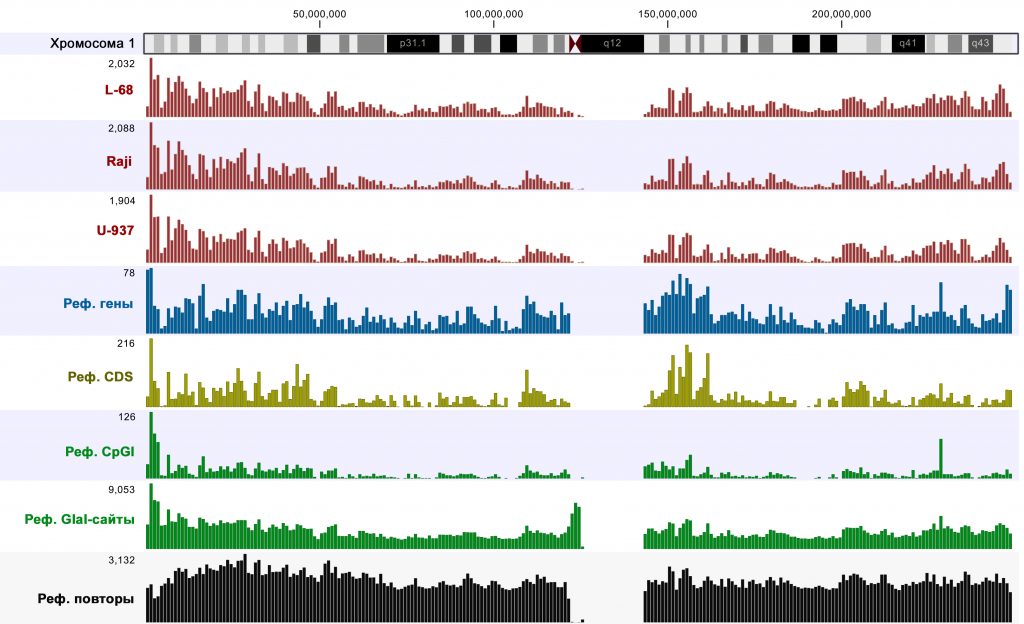
Рис. 1. Диаграммы распределения количеств достоверно покрытых участков и элементов геномной ДНК (CDS – кодирующие участки, CpGI – СpG-островки) для хромосомы 1 (по стандартной аннотации к референсному геному). Вверху показана цитологическая карта хромосомы 1. Цифры слева от диаграмм показывают масштаб (максимальное количество элементов на отрезках по 1 млн п.н.).
Сравнение показало, что, как и в случае предыдущего анализа ДНК Raji [6], распределение надежно покрытых участков значительно варьирует по областям хромосом, но достаточно хорошо коррелирует с таковым для генов и CDS. Таким образом, метод обеспечивает высокую степень покрытия функционально значимых участков генома.
Начальные позиции картированных ридов указывают на места расщепления ДНК эндонуклеазой GlaI и, соответственно, на присутствие метилированных сайтов RCGY в этих положениях. Для дальнейшего анализа была составлена база данных в формате MySQL, включающая 1 696 422 позиции, которые были расщеплены не менее 7 раз для любого из геномов (соответствует значению p<0,005 для достоверно покрытых участков). В базу данных также включены количества зафиксированных расщеплений для каждого генома.
Сравнение полученных позиций с координатами генов из базы данных Gencode показало, что 65% из них располагается в телах генов, но при этом 30% находится в интронных повторах. 4,8% выявленных метилированных сайтов расположены в условных регуляторных участках генов (±500 п.н. от старта транскрипции), а 2,1 % — в CpG-островках. Были подсчитаны числа расщепляемых позиций (с частотой не менее 7 расщеплений) в телах генов, повторах, CpG-островках и условных регуляторных участках для каждого генома и определены процентные доли в общем числе расщеплений (Таблица 2).
Таблица 2
Распределение часто расщепляемых позиций по частям генома (в % от их общего числа для каждого из геномов).| Части генома человека | L-68 | Raji | U-937 |
|---|---|---|---|
| Уникальные участки генов | 29,73 | 31,8 | 32,36 |
| Повторы в межгенных участках | 22,98 | 21,25 | 21,5 |
| Повторы в интронах генов | 35,06 | 35,39 | 33,35 |
| Уникальные межгенные участки | 12,23 | 11,55 | 12,79 |
| Участки ±500 п.н. от стартов транскрипции | 2,79 | 4,4 | 4,58 |
| CpG-островки | 0,79 | 2,73 | 3,02 |
Сравнение данных показывает, что в малигнантных клетках наблюдается существенное возрастание доли расщеплений для CpG-островков (в 3,5 раза для Raji и в 3,8 раза для U-937) и для условных регуляторных участков генов (в 1,6 раза для Raji и U-937) по сравнению с немалигнантными клетками L-68. Это свидетельствует о том, что участки генома, функционально значимые в плане регуляции генной активности, в малигнантных клетках метилированы в большей степени, чем в клетках L-68. Некоторое снижение долей в геномах малигнантных клеток наблюдается для ДНК-повторов, что объясняется их деметилированием при канцерогенезе, хорошо известным по литературным данным [9, 10]. Известно, однако, что существует большое число групп ДНК-повторов различного происхождения, отличающихся по первичной структуре. Дальнейшее сравнение долей расщепленных позиций в классах и семействах самых распространенных повторов показало, что изменения в геномах малигнантных клеточных линий относительно генома L-68 происходят неравномерно.
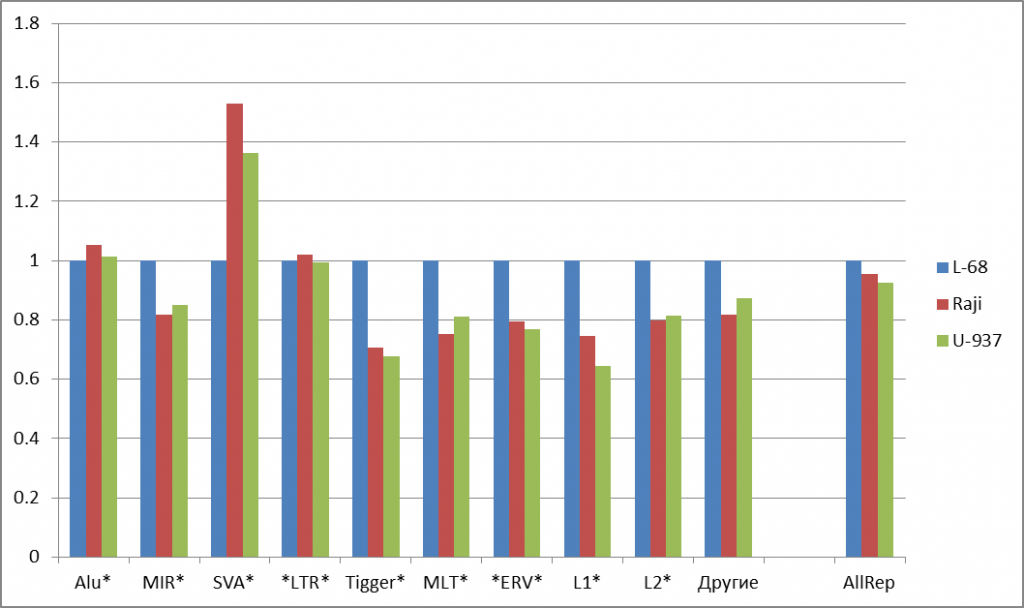
На рисунке 2а представлены диаграммы, показывающие изменения долей расщеплений некоторых распространенных групп ДНК-повторов в геномах малигнантных клетках относительно генома клеточной линии L-68. Из рисунка видно, что для большей части распространенных повторов наблюдается снижение числа расщеплений в геномах малигнантных клеток (на 14-34%). Значительное увеличение доли расщеплений для геномов малигнантных клеток выявлется для SVA-повторов (в 1,53 и 1,36 раз для Raji и U-937, соответственно, по сравнению с L-68). В случае наиболее часто встречающихся Alu- и LTR-повторов различия в долях расщеплений для трех геномов невелики, что несколько нивелирует суммарную картину изменений по всем повторам (последние столбцы на рисунке 2а).
А)
Б)
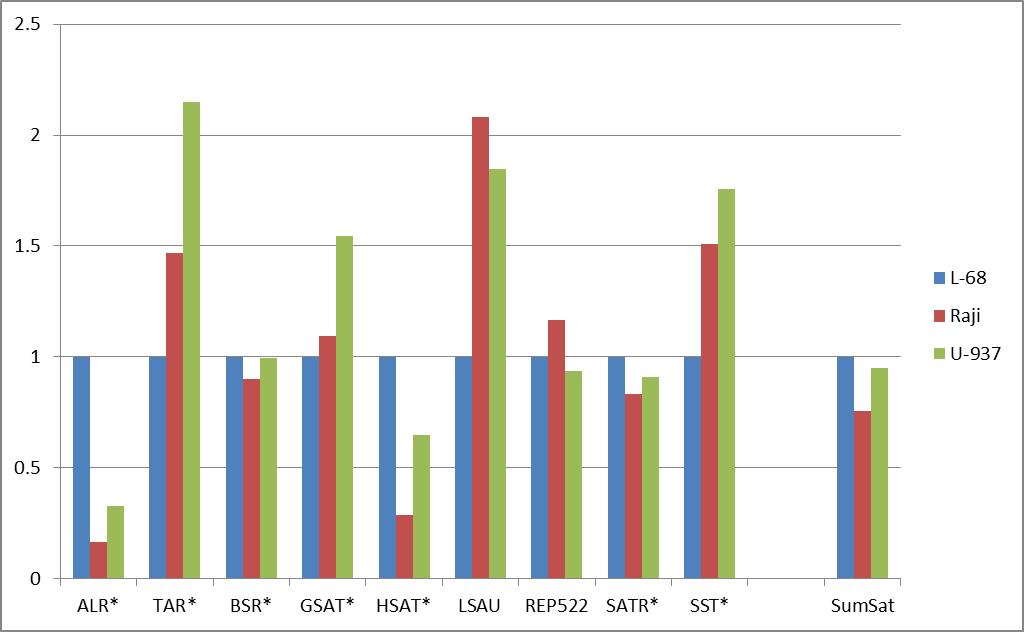
Рис. 2. Изменение долей расщепленных позиций для основных групп повторов (а) и для подгрупп сателлитных повторов (б). Данные нормированы относительно доли расщеплений для генома L-68. Указаны маски для поиска повторов в базе данных, содержащие звездочки (заменяют любые символы в названиях повторов). “AllRep” — сумма всех повторов. “SumSat” — сумма всех сателлитных повторов.
При дальнейшем рассмотрении выяснилось, что внутри групп повторов (для подгрупп) могут наблюдаться существенные различия по долям расщеплений в трех геномах. В качестве примера на рисунке 2б приведено сравнение долей расщеплений для различных видов сателлитной ДНК. В препаратах ДНК из малигнатных клеточных линий наблюдается значительное снижение доли, вносимой ALR- и HSAT-повторами, при существенном возрастании доли у TAR-, LSAU- и SST-повторов, что свидетельствует о неравномерном изменении степени метилирования для разных видов сателлитной ДНК при малигнизации и соответствует ранее опубликованным данным [11-14]. Возможно, что это явление имеет функциональное значение для структурной организации хромосом и аномального метаболизма опухолевых клеток. Но в целом, вопрос неравномерного метилирования различных видов повторов нуждается в подтверждении и дальнейшем изучении, поэтому диагностический потенциал этого явления остается неясным [15].
На следующем этапе работы были определены регуляторные участки генов, наиболее метилированные в трех изучаемых геномах. Для анализа использовались условно регуляторные участки (±500 п.н. от точки старта транскрипции наиболее протяженного транскрипционного варианта). Для каждого гена из базы Gencode подсчитывалось суммарное количество расщеплений ферментом GlaI на условно регуляторном участке по каждому геному. Из полученной выборки для каждого генома были исключены гены, содержащие менее 7 расщеплений в своем условном регуляторном участке. Полученные списки генов были ранжированы по суммарному количеству расщеплений и проанализированы с помощью сервиса Venn Diagramm (http://bioinformatics.psb.ugent.be/webtools/Venn/). Это позволило определить те гены, которые являются общими для трех геномов, для каждой пары геномов или являются специфическими лишь для одного генома. Количество общих и уникальных генов с метилированными условно регуляторными участками приведено на рисунке 3.
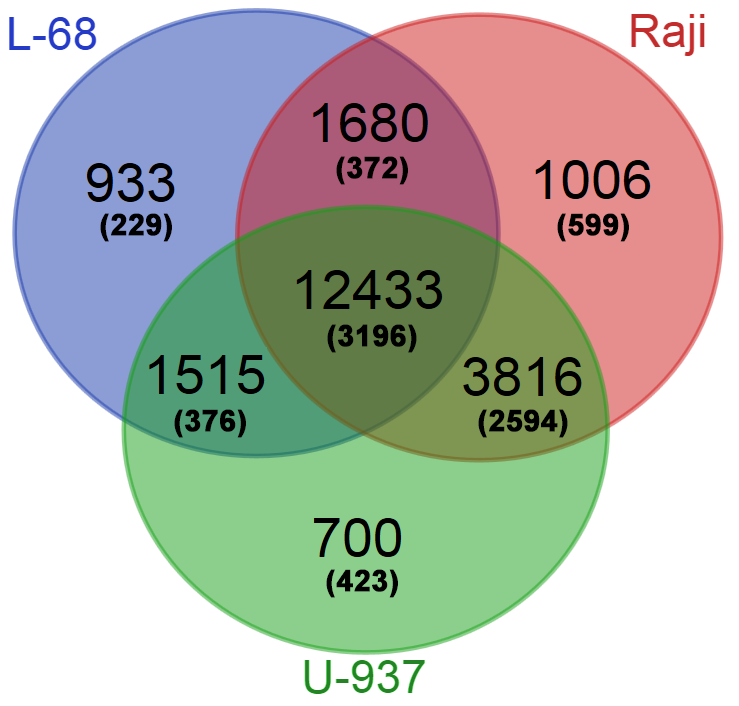
Рис. 3. Общие и уникальные гены с метилированными условно регуляторными участками для трех геномов. В скобках указано число генов, кодирующих белки, которые могут быть идентифицированы по классификации Gene Ontology.
Часть из генов Gencode кодирует белки и может быть отнесена к какой-либо из категорий по классификации Gene Ontology [16]. Для определения того, в каких именно биологических процессах участвуют гены с метилированными регуляторными участками провели GO-анализ списка 2594 генов, общих только для геномов малигнантных клеточных линий, и списка 229 генов, свойственных только геному клеточной линии L-68. Установление основных категорий проводили, используя сервис GOrilla [17]. Результаты проведенного анализа представлены в таблице 3 (указаны лишь первые семь категорий).
Таблица 3
Основные категории GO для биологических процессов, для которых наблюдаются существенные отклонения от ожидаемых количеств генов в анализируемых списках.| Термин GO | Описание биологического процесса | Значение P | |
|---|---|---|---|
| Метилированные гены, общие только для геномов малигнантных клеток | GO:0030154 | cell differentiation | 2.41E-07 |
| GO:0032502 | developmental process | 1.12E-06 | |
| GO:0048869 | cellular developmental process | 1.66E-06 | |
| GO:0044767 | single-organism developmental process | 2.36E-06 | |
| GO:0048856 | anatomical structure development | 2.59E-06 | |
| GO:0010468 | regulation of gene expression | 7.15E-06 | |
| GO:0051240 | positive regulation of multicellular organismal process | 1.94E-05 | |
| Метилированные гены, уникальные для генома клеточной линии L-68 | GO:0071704 | organic substance metabolic process | 6.38E-06 |
| GO:0044238 | primary metabolic process | 8.36E-06 | |
| GO:0008152 | metabolic process | 9.76E-05 | |
| GO:0006725 | cellular aromatic compound metabolic process | 1.84E-04 | |
| GO:1901360 | organic cyclic compound metabolic process | 1.84E-04 | |
| GO:0090304 | nucleic acid metabolic process | 2.18E-04 | |
| GO:0006139 | nucleobase-containing compound metabolic process | 2.47E-04 |
Из таблицы следует, что метилирование в геномах малигнантных клеток затрагивает в первую очередь регуляторные участки генов, участвующих в процессах развития организма и клеточной дифференцировки. Это хорошо согласуется с потерей специализации, свойственной опухолевым клеткам. Для генома немалигнантной клеточной линии L-68 наблюдается метилирование генов различных метаболических процессов, что, вероятно, связано с поддержанием пониженного уровня активности дифференцированных клеток. В целом, эти данные соответствуют современным представлениям о различиях между малигнантными и нормальными клетками.
Для извлечения данных из созданной базы нами написано программное обеспечение «MGenome Browser» (http://mbrowser.sibenzyme.com, рисунок 4). Этот сервис позволяет искать позиции расщепления ферментом GlaI в геномах трех клеточных линий по названиям генов или по референсным координатам участков генома. Поддерживается автозаполнение имен генов, а также выбор определенных клеточных линий и минимальной частоты детектированных расщеплений в сайтах RCGY на выбранном участке. База данных в будущем может быть расширена по мере проведения секвенирования других геномов человека.
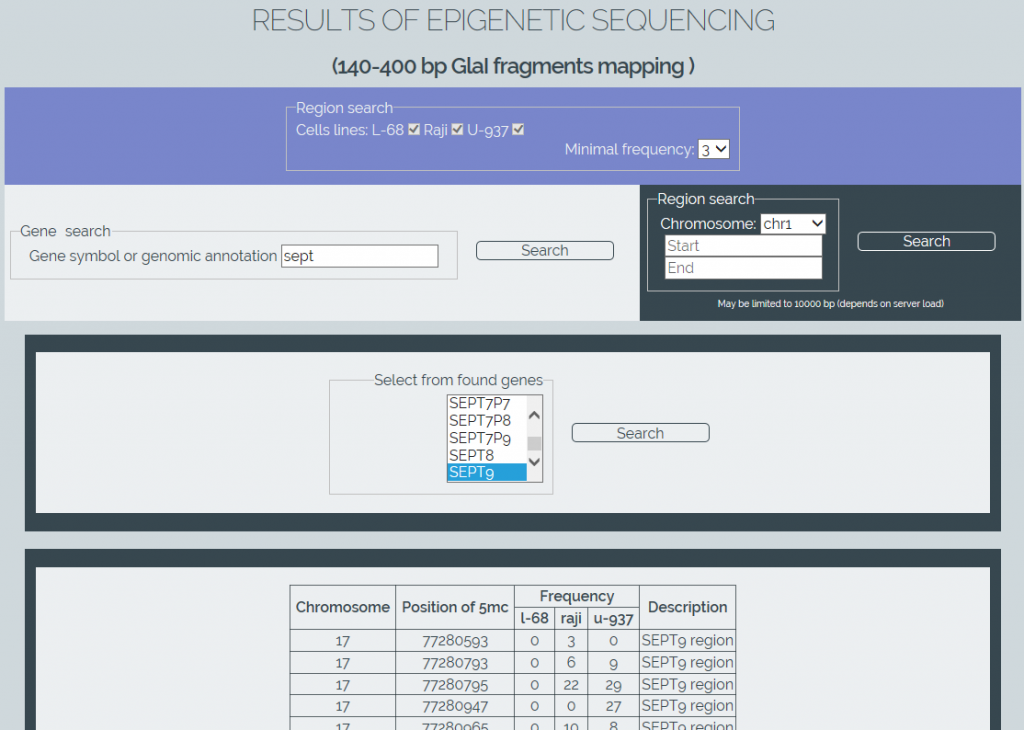
Рис. 4. Скриншот веб-страницы сервиса «MGenome Browser».
ОБСУЖДЕНИЕ
Как и другие методы анализа метилирования с помощью метилзависимых или метилчувствительных эндонуклеаз [18-21], метод секвенирования GlaI-фрагментов не позволяет выявлять все метилированные цитозиновые основания генома, так как расщепление может происходить лишь по сайтам узнавания ферментов. Другим ограничением разрабатываемого метода является возможность секвенирования фрагментов лишь определенного диапазона длин, связанная с принципами работы современных секвенирующих машин. Например, при гидролизе участков CpG-островков, насыщенных метилированными сайтами RCGY, образуются слишком короткие фрагменты для анализа. Однако, как было показано, метилирование в менее GC-богатых участках генома, таких как «берега CpG-островков» и отдаленных энхансерах, также важно для регуляции генной активности и может использоваться для различения опухолевых и нормальных клеток [22, 23]. Полученные нами результаты указывают на то, что число выявляемых метилированных сайтов в геноме достаточно велико и превышает число, определяемое с помощью ДНК-чиповых технологий (например, с помощью Illumina Infinium). Это дает возможность проводить сравнительный анализ с целью выявления позиций, перспективных в качестве эпигенетических маркеров заболеваний. Важным преимуществом нового метода является его простота по сравнению с методами, основанными на аффинном связывании метилированных участков и/или бисульфитной конверсии, которые все еще трудоемки и дорогостоящи [24, 25].
ВЫВОДЫ
В настоящей работе представлены результаты картирования метилированных сайтов RCGY в геномах двух малигнантных клеточных линий и контрольной клеточной линии фибробластов легких. Проведенный анализ выявил существенные отличия по метилированию генов, повторов, CpG-островков между тремя геномами. Обнаружены различия по степени метилирования различных групп повторов, которые, возможно, могут быть использованы в диагностических целях при условии разработки достаточно чувствительных методов анализа. Создана база данных, содержащая результаты картирования метилированных сайтов в геномах. Полученные данные по метилированию регуляторных участков различных генов, сопоставленные с ранее опубликованными данными, могут быть использованы для поиска маркеров метилирования для диагностики онкологических и некоторых других заболеваний. Таким образом, разработанный простой и нетрудоемкий метод картирования метилированных сайтов R(5mC)GY в геномах может быть использован на практике.
СПИСОК ЛИТЕРАТУРЫ
[1] Tollefsbol TO, Ed. Epigenetics in human disease. Amsterdam, New York: Academic Press; 2012.
[2] Brookes E, Shi Y. Diverse epigenetic mechanisms of human disease. Annu Rev Genet. 2014;48:237-68.
[3] García-Giménez JL, Ed. Epigenetic biomarkers and diagnostics. London: Academic Press; 2015.
[4] Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191-203.
[5] Fouse SD, Nagarajan RP, Costello JF. Genome-scale DNA methylation analysis. Epigenomics. 2010;2:105–17.
[6] Abdurashitov MA, Tomilov VN, Gonchar DA, Kuznetsov VV, Degtyarev SK. Mapping of R(5mC)GY sites in the genome of human malignant cell line Raji. Biol Med (Aligarh). 2015;7:BM-135-15.
[7] Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigó R, Hubbard TJ. GENCODE: the reference human genome annotation for the ENCODE project. Genome research. 2012;22:1760-74.
[8] Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, Heitner S, Harte RA, Haeussler M, Guruvadoo L, Fujita PA, Eisenhart C, Diekhans M, Clawson H, Casper J, Barber GP, Haussler D, Kuhn RM, Kent WJ. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016;44(D1):D717-25.
[9] Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400-13.
[10] Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138-62.
[11] Choi SH, Worswick S, Byun HM, Shear T, Soussa JC, Wolff EM, Douer D, Garcia-Manero G, Liang G, Yang AS. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int J Cancer. 2009;125:723-9.
[12] Nishiyama R, Qi L, Tsumagari K, Weissbecker K, Dubeau L, Champagne M, Sikka S, Nagai H, Ehrlich M. A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol Ther. 2005;4:440-8.
[13] Nishiyama R, Qi L, Lacey M, Ehrlich M. Both hypomethylation and hypermethylation in a 0.2-kb region of a DNA repeat in cancer. Mol Cancer Res. 2005;3:617-26.
[14] Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014;15:583.
[15] Ross JP, Rand KN, Molloy PL. Hypomethylation of repeated DNA sequences in cancer. Epigenomics. 2010;2:245-69.
[16] The Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucl Acids Res. 2015;43(Database issue):D1049-56.
[17] Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009, 10:48.
[18] Fouse SD, Nagarajan RO, Costello JF. Genome-scale DNA methylation analysis. Epigenomics. 2010;2:105-17.
[19] Harrison A, Parle-McDermott A. DNA methylation: a timeline of methods and applications. Front Genet. 2011;2:74.
[20] Yong WS, Hsu FM, Chen PY. Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016;9:26.
[21] Cohen-Karni D, Xu D, Apone L, Fomenkov A, Sun Z, Davis PJ, Kinney SR, Yamada-Mabuchi M, Xu SY, Davis T, Pradhan S, Roberts RJ, Zheng Y. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc Natl Acad Sci U S A. 2011;108:11040-5.
[22] Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178-86.
[23] Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11-3.
[24] Umer M, Herceg Z. Deciphering the epigenetic code: an overview of DNA methylation analysis methods. Antioxid Redox Signal. 2013;18:1972-86.
[25] Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191-203.
