Comparative analysis of RCGY sites methylation in three human cell lines
Abdurashitov M.A.1,*, Tomilov V.N.1, Gonchar D.A.1, Snezhkina A.V.2, Krasnov G.S.2, Kudryavtseva A.V.2,3, Degtyarev S.Kh.1
1 SibEnzyme Ltd., Novosibirsk 630117, Russia
2 Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow 119991, Russia
3 National Medical Research Radiological Centre, Ministry of Healthcare of the Russian Federation, Moscow 125284, Russia
* Corresponding author: Abdurashimov M, Ph.D., SibEnzyme, 2/12 Ak.Timakov Str., Novosibirsk 630117, Russia; Tel: -7 383 3334991; Fax: -7 383 3336853; E-mail: abd@sibenzyme.ru
DNA methylation in human genome is important for the cells specialization and functioning. An abnormal methylation of the regulation regions of some genes may cause the genes silencing and this phenomenon is often detected in cancer cells. Determination of differences of the genome-wide methylation in normal and tumor cells is useful for understanding the carcinogenesis process and for development of new methods of epigenetic diagnostics. The positions of methylated RCGY sites in the genomes of Raji, U-937 and L68 human cell lines have been determined using the previously developed method of massive parallel sequencing of Glal fragments. A comparison of the obtained data has revealed significant differences in methylation of CpG islands, putative regulatory regions and some repetitive DNA families between genomes of malignant and non-malignant cells. GO enrichment analysis of genes with highly methylated regulatory regions has shown the possible metabolic processes, which may be affected epigenetically in carcinogenesis. The new method allows to determine positions of many modified cytosine bases in the genomes and may be a simple alternative to the existing methods of genome-wide methylation analysis.
Keywords: DNA methylation, epigenomics, next-generation sequencing, methyl-directed DNA endonucleases
DOI: 10.26213/SE.2019.76.40116
Citation: M.A. Abdurashitov, V.N. Tomilov, D.A. Gonchar, A.V. Snezhkina, G.S. Krasnov, A.V. Kudryavtseva, S.Kh. Degtyarev (2019) Comparative analysis of RCGY sites methylation in three human cell lines. Epigenetic DNA diagnoctics, vol.2019(1), DOI: 10.26213/SE.2019. 76.40116
Introduction
Epigenetic alterations play an essential role in the regulation of gene activity. Aberrant methylation of the genomic regulatory regions may change normal functioning of cells and often accompanies a number of human disorders including cancer. Thus, methylation markers have diagnostic value and have attracted attention of many biomedical researchers [1-3). But the selection of the most reliable biomarkers requires an analysis of the methylation data obtained from many genomes. The genome methylation data are now available due to application of next generation sequencing (NGS) technologies, however the existing methodologies need improvements to make them cost-effective and less laborious (4, 5).
Earlier we proposed a simple method of methylated RCGY sites determination in the whole genome. The method is based on genomic DNA cleavage with site-specific methyl-directed DNA-endonuclease Glal followed by massive parallel sequencing of obtained fragments (140-400 bp in length). The results of the trial experiment using lllumina MiSeq have allowed us to determine more than 1 million methylated sites R(5mC)GY in DNA from the Raji cell line. The obtained data were compared to the results of PCR analysis of several regulatory regions and showed a good correspondence to them [6].
In this work, we present a comparative analysis of R(5mC)GY sites revealed in the genomes of three cell lines using the same approach. Non-malignant L-68 cell line and malignant cell line U-937 have been selected for study. Raji genomic DNA fragments has been resequenced to confirm the reproducibility of the method. To increase reliability of the data, we have chosen the lllumina Genome Analyzer llx (GAllx) system for sequencing, which allows us to obtain more reads in comparison to MiSeq.
Materials and Methods
Cells of three lines were obtained from the “Collection of Microorganisms” Department of The State Research Center of Virology and Biotechnology “Vector” (Koltsovo, Novosibirsk region, Russia). Genomic DNA isolation has been performed using standard phenol chloroform extraction from the cell lysates. Glal enzyme was supplied by SibEnzyme Ltd. (Novosibirsk, Russia) and enzymatic reactions were performed as recommended by the manufacturer. Products of DNA hydrolysis were separated by electrophoresis in 1x Tris-acetate buffer using 1.4% agarose gel. Glal DNA fragments 140-400 bp in length were cut from gel and purified using the “Cyt 202” kit from Cytokin Ltd. (St. Petersburg, Russia). DNA libraries for sequencing on the lllumina Genome Analyzer llx instrument were prepared using standard protocol provided by the manufacturer. 75 bp from both ends of the fragments were sequenced.
The obtained reads were filtered for rejection of sequences that did not contain dinucleotide GY at 5′ end, and therefore, were not products of Glal digestion. Additionally, the reads containing more than 25 undetermined nucleotides were excluded from the final results.
The reference human genome sequence GRCh38.p3 and annotations for genes (Gencode release 23 [7]), coding DNA sequences (CDSs), CpG islands and DNA repeats were obtained from UCSC Genome Bioinformatics site [8]. The mapping of reads on the reference genomic sequence, as well as visualization of mapped reads and annotations, were performed using CLC Genomics Workbench software (Qiagen Aarhus A/S, Aarhus, Denmark).
Results
Approximately 100 mln reads were obtained for each studied genome after sequencing using the Genome Analyzer llx. About 2/3 of the total reads passed through filtration and mapping steps. The statistical data on the sequencing results are given in Table 1.
Table 1
Statistics on the sequencing of 140-400 bp GiaI fragments for genomes of three cell lines| Cell line | Total reads | Filtered reads | Mapped reads | Covered part of the reference genome | Average coverage (not including non covered regions) |
|---|---|---|---|---|---|
| L-68 | 100 362 080 | 79 626 274 | 72 144 184 | 12% | 14,7 |
| Raji | 94 526 060 | 70 252 160 | 61 364 793 | 10% | 14,9 |
| U-937 | 103 712 902 | 66 477 816 | 59 179 479 | 10% | 14,24 |
The starting coordinates of the mapped reads show the position of GY dinucleotide from methylated RCGY sites. All these coordinates were extracted from the mapping results for each genome to the SQL database. The numbers of reads which starts from these coordinates were also added to the database for indication of cleavage events at each revealed point.
To confirm the reproducibility of the method, we compared the newly obtained data for the Raji genome to data obtained previously for the same genome using MiSeq for sequencing [6]. There are approximately 7.3 mln RCGY sites in the reference genome and more than 3.5 mln of these were detected in the experiment. However, combining of MiSeq data with GAllx data increased the Raji methylation database only by 1.8% and almost all additional positions had very low frequency of cleavage (1 to 3). This indicates that the main core of revealed methylated positions did not change significantly in the two performed experiments.
Visualization of mapping results and comparison to reference genome annotations using CLC Genomics Workbench software showed that highly covered regions (p-value <0.005) were mainly located in the genomic parts enriched with genes, CDSs and CpG islands. An example of such analysis for chromosome 1 is given in Figure 1. It should be mentioned that an almost identical pattern was observed for Raji DNA after MiSeq sequencing [6], thus, also confirming reproducibility of the method.
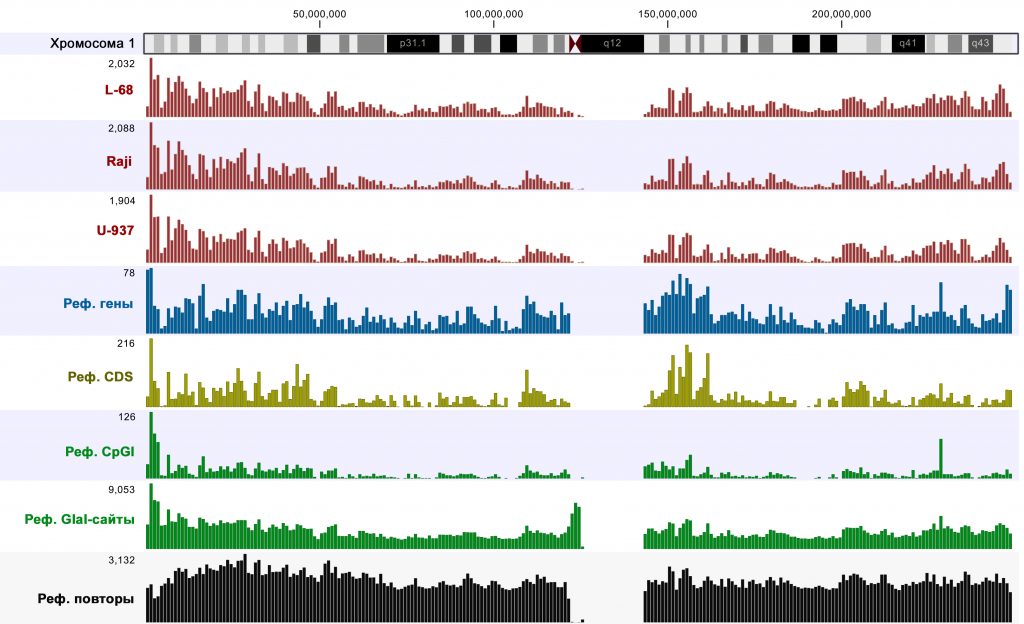
Figure 1: Amounts of highly covered regions, genes, CDSs, CpG islands, RCGY sites and DNA repeats in 1 Mbp segments of the human chromosome1. Cytogenetic ideogram is shown at top. Scales are shown by numbers in top left corner of diagrams
For further analysis, we excluded from the database all cleavage positions that were detected less than 7 times (in correspondence to a selected p-value for highly covered genomic regions) in all three genomes. This allowed us to select only reliable values reflecting high methylation level at the certain positions in at least one genome. The zip-archived CSV version of the final database can be found in Supplementary Materials. It contains 1,696,422 positions with corresponding numbers of detected cleavage events for each genome.
The comparison of these positions to the reference genome annotations showed that -65% of the revealed sites were located within gene bodies, however -30% of them were in intronic repeats. 2.1% of the detected cleavage positions were located in the CpG islands, whereas 4.8% were located in the putative gene regulatory regions (±500 bp from the starts of transcription).
In order to determine whether there is a difference in cleavage frequencies of genomic elements between the three genomes, we calculated the total numbers of positions with more than 7 cleavages for each genome. Table 2 shows the results of this analysis
Table 2
Distribution of cleavage events in the genomic elements (In percents to total number of cleaved positions for whole genome, the positions with a number of cleavage events <7 were not taken into account.)| Genomic regions | L-68 | Raji | U-937 |
|---|---|---|---|
| Unique parts of genes | 29,73 | 31,8 | 32,36 |
| Repetitive DNA outside genes | 22,98 | 21,25 | 21,5 |
| Repetitive DNA in intrans | 35,06 | 35,39 | 33,35 |
| Unique DNA outside genes | 12,23 | 11,55 | 12,79 |
| Regions ±500 bp from transcription starts | 2,79 | 4,4 | 4,58 |
| CpG-islands | 0,79 | 2,73 | 3,02 |
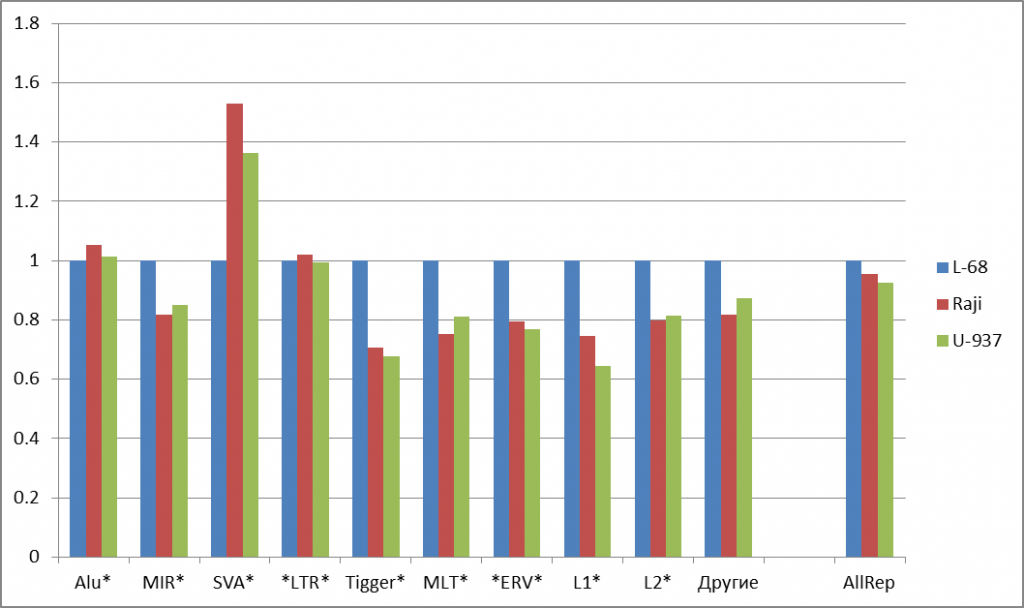
The most considerable changes in methylation levels for genomes of malignant cells in comparison to the genome of non-malignant cell line L-68 is observed in the CpG islands (a 3.5 and 3.8 fold increase for Raji and U-937, correspondingly). Putative regulatory regions of genes show less significant changes (a 1.6 fold increase for Raji and U-937).Thus, these data show that many regulation elements of the genome, which are functionally important for gene activity, are methylated in the studied tumor cells. Comparison of methylation in gene bodies doesn’t give a clear picture of possible epigenetic alterations in carcinogenesis. DNA repeats are less methylated in the studied malignant cells than in non-malignant L-68 cells, and this fact correlates with the published data on hypomethylation of repetitive DNA in the genomes of tumor cells [9, 1O]. However, there are many groups of DNA repeats of variable primary structure, and little is known about their epigenetic distinctions. So, it was of interest to compare the levels of methylation for groups of repeats in this study. Figure 2a shows the significant changes in the content of summarized cleavage positions in some abundant human DNA repeats in the malignant cell line relative to those in the non-malignant cell line. For most of examined repetitive groups, a decrease in methylation level by 14-34% was observed for malignant DNA repeats. However, no significant changes in methylation levels for Alu and LTR repeats were detected, while SVA repeats showed 53% and 36% increase for Raji and U-937 genomes, respectively, compared to the L-68 genome.
a)
b)
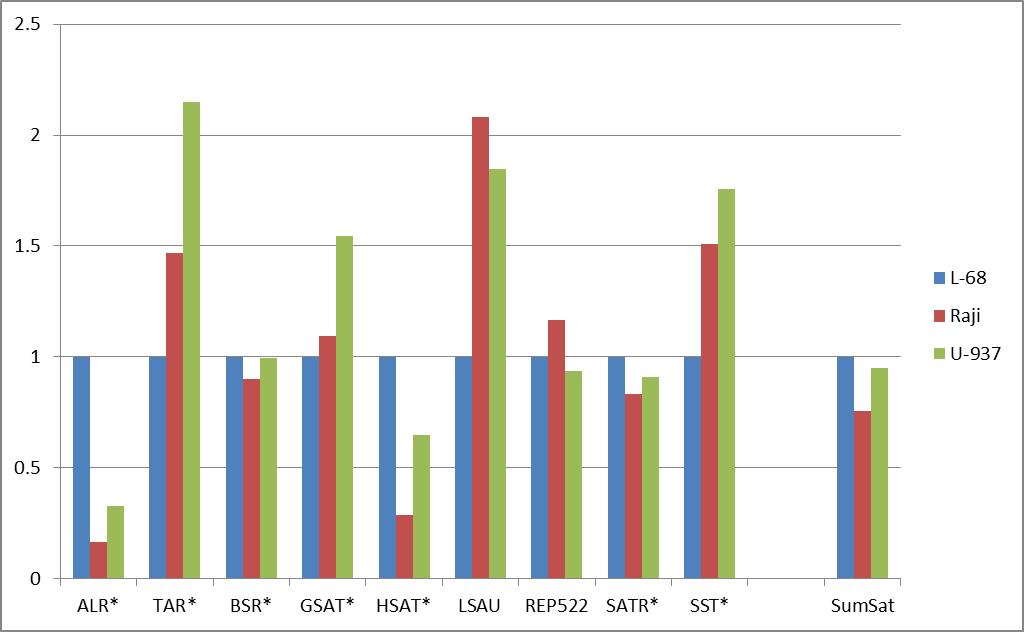
Figure 2: Fold changes of the total numbers of cleavage events for abundant groups of DNA repeats (a) and for satellite DNA repeats (b). Normalization to L-68 genome was used. The masks used for repetitive DNA groups search in the database are shown at bottom. AllRep – a sum of all repeats, SumSat – a sum of satellite repeats.
Satellite DNA, though considered as one class of DNA repeats, actually incorporates heterogeneous groups of tandem repeats not related evolutionarily. Therefore, satellite repeats were analyzed separately and comparison of their methylation showed an unusual variability (Figure 2b). ALR and HSAT repeats showed a significant decrease of methylation level in contrast to TAR, LSAU and SST repeats demonstrated an increased levels of methylation in malignant cells compared to non-malignant cells. Probably the inequality of methylation levels for different repeats may play some functional role for structural organization of chromosomes in malignant cells, thus affecting normal cell metabolism. Our results support previously published data on the variability of methylation of different tandem repeats in cancer [11]. For example, LSAU and SST repeats are partially included in 0424 and NBL2 complex repetitive sequences, respectively, and aberrant methylation levels, which have been shown in tumor cells in a number of studies [11- 13]. The reported changes in the expression levels of the certain repetitive DNA groups in cancer cells may be explained by abnormal methylation of these DNA regions [14]. However, in general, the epigenetic alterations of repetitive DNA are not well studied and require further clarification. The previously obtained results indicate that different cancers may show non-similar patterns of methylation of DNA repeats, so the diagnostic potential of this phenomenon is still unclear [15].
The obtained database of Glal cleavage positions was also used for determination of the genes with high methylation levels in their putative regulatory regions. For each gene from Gencode, a total number of cleavage events in the region ±500 bp from the gene start were calculated for each genome. Genes with a total number of cleavages in the putative regulatory regions less than 7 were excluded from the final gene lists. The three obtained gene lists were compared online using Venn Diagramm tool (http://bioinformatics.psb.ugent.be/webtoolsNenn/). Figure 3 shows amounts of common and unique methylated genes in the studied genomes.
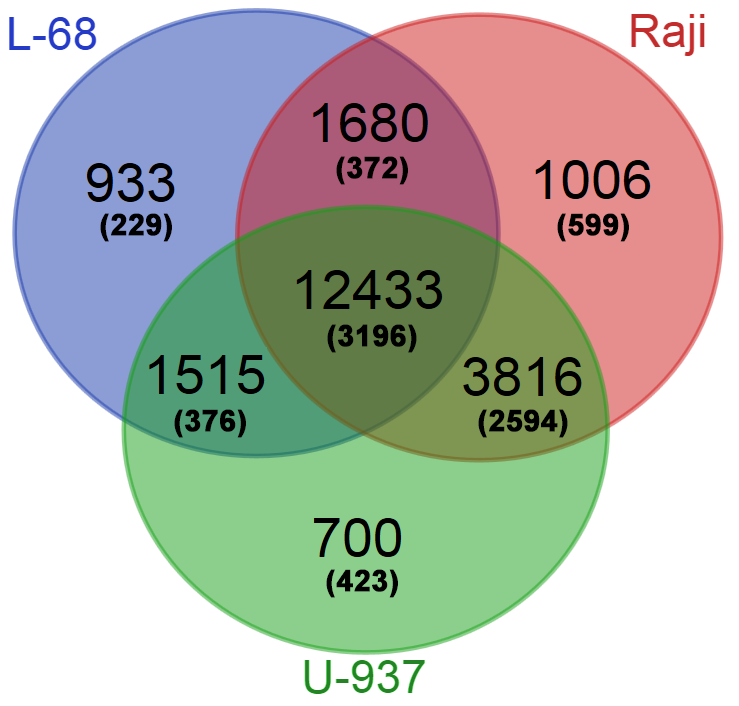
Figure 3: Numbers of common and unique genes with methylated putative regulatory regions in the studied genomes
Part of Gencode genes encode proteins with known functions and may be categorized using Gene Ontology classification [16]. To reveal the biological processes which may be affected by methylation, we performed GO enrichment analysis for the genes common for malignant cells and for the genes which were methylated in the non-malignant L-68 cell line. GOrilla service was used for determination of main GO terms over-represented in the gene lists [17]. The obtained data are presented in Table 3.
GO analysis of genes methylated in both malignant cells showed a significant enrichment in terms related to cell differentiation and processes of the organism growth. This may reflect a loss of cell specialization characteristic for malignant cells. In L-68 genome we observe a methylation in genes of different processes of metabolism.
Table 3
The main GO terms obtained for genes with methylated regulatory regions in Raji and U-937 genomes| GO terms | Description | P value | |
|---|---|---|---|
| Methylated genes common only to the genomes of malignant cells | GO:0030154 | cell differentiation | 2.41E-07 |
| GO:0032502 | developmental process | 1.12E-06 | |
| GO:0048869 | cellular developmental process | 1.66E-06 | |
| GO:0044767 | single-organism developmental process | 2.36E-06 | |
| GO:0048856 | anatomical structure development | 2.59E-06 | |
| GO:0010468 | regulation of gene expression | 7.15E-06 | |
| GO:0051240 | positive regulation of multicellular organismal process | 1.94E-05 | |
| Methylated genes unique to the L-68 cell line genome | GO:0071704 | organic substance metabolic process | 6.38E-06 |
| GO:0044238 | primary metabolic process | 8.36E-06 | |
| GO:0008152 | metabolic process | 9.76E-05 | |
| GO:0006725 | cellular aromatic compound metabolic process | 1.84E-04 | |
| GO:1901360 | organic cyclic compound metabolic process | 1.84E-04 | |
| GO:0090304 | nucleic acid metabolic process | 2.18E-04 | |
| GO:0006139 | nucleobase-containing compound metabolic process | 2.47E-04 |
* Child GO levels are indicated using additional spaces before term designations
We also have developed software for online retrieval of the results from the obtained methylation database (MGenome Browser, http://mbrowser.sibenzyme.com).MGenome Browser supports two ways of locating the methylated cytosines: by linking to gene locations in the genome or by directly searching certain regions of chromosomes. The software also supports a gene name autocompletion. It also allows the user to select one or more cell lines to perform a search and to set a minimum frequency of the detected Glal cleavages to refine a search (Figure 4). Output results are displayed as table data.
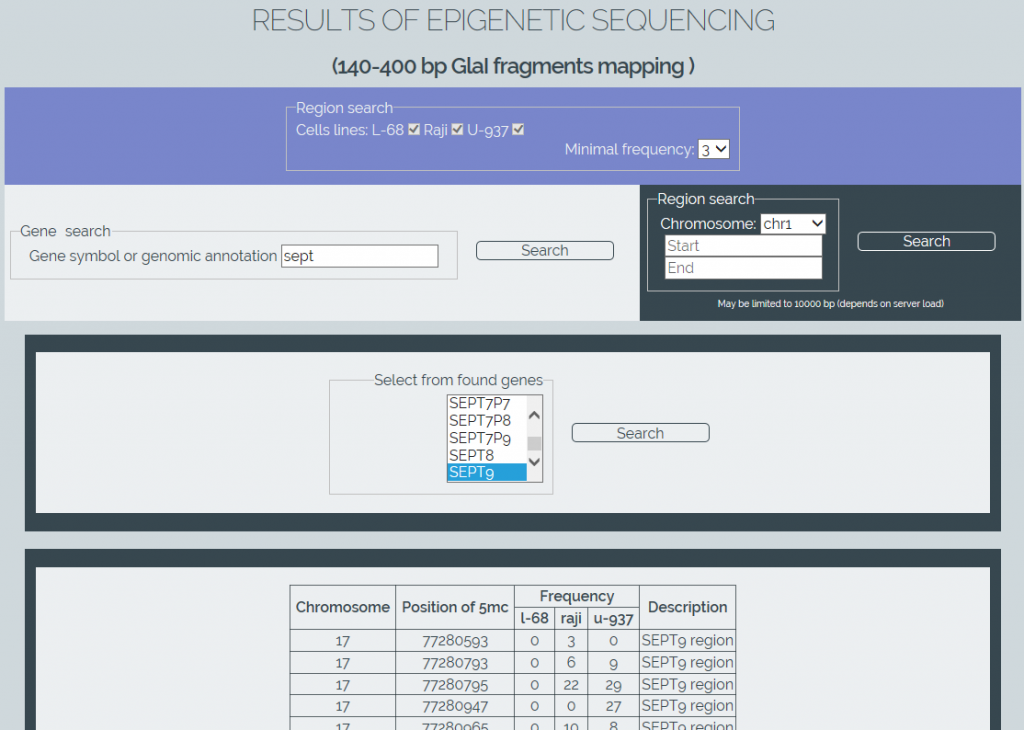
Figure 4: Screenshot of MGenome Browser webpage
Discussion
Like other methods based on usage of methylation-sensitive or methylation-dependent endonucleases [18-21], the sequencing of Glal fragments does not allow to reveal all methylated cytosine residues in the genome. Only recognition sites of the enzymes may be analyzed by these approachs. Other limitation of the described method is related to the DNA fragment length restrictions for modern NGS devices. So, only part of the total genomic fragments pool is suitable for sequencing thus enhancing the data incompleteness. For example, the high density of the methylated RCGY sites in the middle of CpG island may lead to formation of small fragments after Glal hydrolysis which are undetectable using sequencing of the selected 140-400 bp fragment pool. However, it was shown that methylation in less GC-rich regions such as CpG islands “shores” and distal control regions (enhancers) are also important for gene regulation and may be used to distinguishnormal and tumor cells [22, 23]. Our results show that the number of the frequently methylated bases which may be revealed by the new method is rather high. This allows to carry out a comparative analysis of the data from different genomes and to point positions which may be interesting for use as diagnostic markers. The serious advantage of our method is its simplicity in comparison to enrichment- or bisulfite-based methods of genome-wide epigenetic analysis which are much more laborious and less cost-effective [24, 25]. The method requires only one-step preliminary DNA hydrolysis using Glal enzyme which produces blunt-ended fragments suitable for further processing according to the NGS device manufacturer’s protocol.
Conclusions
In this work we have applied the developed method of Glal fragments sequencing for epigenetic study of three genomes. The analysis showed a significant difference in methylation of CpG islands, some groups of DNA repeats and putative regulatory regions of genes.
Thus, the applicability of the method for the epimarkers search was confirmed, and its simplicity allows routine use. Taking into account the variability of different types of cancers, more genomes must be included in the analysis to get the methylation data by the proposed method. This will allow us to find epimarkers specific for certain types of disease and to develop the corresponding diagnostic panels.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
This work was supported by the grant from the Ministry of Education and Science of Russian Federation according to the Agreement No.14.604.21.0102 of 05.08.2014 (unique identifier RFMEFl60414X0102) concluded within the Federal Targeted Program “Research and Development in the Priority Areas of Directions of the Russian Scientific and Technological Complex for 2014-2020”. Part of this work (DNA libraries preparation and sequencing) was performed using the equipment of EIMB RAS “GenomeCenter”. We thank Prof. Nikolay Yankovsky (The Vavilov Institute of General Genetics) for idea of Glal usage in genomic sequencing and for useful discussion.
References
- Tollefsbol TO, Ed. Epigeneitcs in human disease. Amsterdam, New York: Academic Press; 2012.
- Brookes E, Shi Y. Diverse epigenetic mechanisms of human disease. Annu Rev Genet. 2014;48:237-68.
- Garcia-Gimenez JL, Ed. Epigenetic biomarkers and diagnostics. London: Academic Press; 2015.
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191-203.
- Fouse SD, Nagarajan RP, Costello JF. Genome-scale DNA methylation analysis. Epigenomics. 2010;2:105- 17.
- Abdurashitov MA, Tomilov VN, Gonchar DA, Kuznetsov W, Degtyarev SK. Mapping of R(5mC)GY sites in the genome of human malignant cell line Raji. Biol Med (Aligarh). 2015;7:BM-135-15.
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guig6 R, Hubbard TJ. GENCODE: the reference human genome annotation for the ENCODE project. Genome research. 2012;22:1760-74.
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, Heitner S, Harte RA, Haeussler M, Guruvadoo L, Fujita PA, Eisenhart C, Diekhans M, Clawson H, Casper J, Barber GP, Haussler D, Kuhn RM, Kent WJ. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016;44(D1):D717-25.
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400-13.
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138-
- Choi SH, Worswick S, Byun HM, Shear T, Soussa JC, Wolff EM, Dauer D, Garcia-Manero G, Liang G, Yang AS. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int J Cance.r 2009;125:723-9.
- Nishiyama R, Qi L, Tsumagari K, Weissbecker K, Dubeau L, Champagne M, Sikka S, Nagai H, Ehrlich A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol Ther. 2005;4:440-8.
- Nishiyama R, Qi L, Lacey M, Ehrlich M. Both hypomethylation and hypermethylationin a 0.2-kb region of a DNA repeat in cancer. Mol Cancer Res. 2005;3:617-26.
- Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014;15:583.
- Ross JP, Rand KN, Molloy PL. Hypomethylation of repeated DNA sequences in cancer. Epigenomics. 2010;2:245-69.
- The Gene Ontology Consortium. Gene Ontology Consortium: going Nucl Acids Res. 2015;43(Database issue):D1049-56.
- Eden E, Navan R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009, 10:
- Fouse SD, Nagarajan RO, Costello JF. Genome-scale DNA methylation analysis. Epi 2010;2:105-17.
- Harrison A, Parle-McDermott A. DNA methylation: a timeline of methods and applications. Front Genet. 2011;2:
- Yong WS, Hsu FM, Chen PY. Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016;9:26.
- Cohen-Kami D, Xu D, Apone L, Fomenkov A, Sun Z, Davis PJ, Kinney SR, Yamada-Mabuchi M, Xu SY, Davis T, Pradhan S, Roberts RJ, Zheng Y. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc Natl Acad Sci US 2011;108:11040-5.
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue specific CpG island shores. Nat Genet. 2009;41:178-86.
- Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11-3.
