GlaI-PCR analysis of methylation of ACGC site in Chr11: 65647266 in DNA samples from the blood cells of healthy donors and early stage breast cancers
A.G. Akishev1*, N.A.Netesova2, M.A. Abdurashitov1, I.V. Vihlyanov3, M.K. Nikitin3, A.B. Karpov4, S.Kh. Degtyarev1
1 SibEnzyme, Novosibirsk
2 Epigenlab, Novosibirsk
3Altai Regional Oncology Center, 656049, Altai region, Barnaul
4Seversk Biophysical Research Centre, Seversk, Tomsk region
* corresponding author: Alexander Akishev, SibEnzyme, 2/12 Ak.Timakov Str., Novosibirsk 630117, Russia; Tel: +7 383 3334991; Fax: +7 383 3336853; E-mail: aki@sibenzyme.ru
Methylation of ACGC site in position Chr11: 65647266 (according to GRCh38 PrimaryAssembly) in DNA preparations from blood cells of healthy donors and early stage breast cancers was studied with GlaI-PCR assay. The work includes a) preparation of light fraction of the blood cells, b) isolation of genomic DNA, c) determination of genomic DNA concentration by real-time PCR, d) determination of concentration of unmethylated ACGC site in position Chr11: 65647266 with GlaI-PCR assay and e) calculation of percent of DNA molecules with unmethylated ACGC site in position Chr11: 65647266.
GLAD-PCR analysis showed that in more than 82% of donors, the level of ACGC methylation in the SIPA1 gene is 3% or less, while in approximately 70% of patients with breast cancer, the level of ACGC site methylation in SIPA1 gene is in the range of 3.2 to 6.4%. These data suggest that at early stages of breast cancer, demethylation of A(5mC)GC site in the SIPA1 gene at position 65647266 takes place in 2/3 of patients. The data of this work may be used for a development of PCR test-system for exclusion of diagnosis “early stage breast cancer”. GlaI-PCR assay doesn’t require special and expensive equipment. The study may be carried out in a standard PCR laboratory when a blood analysis is taken place.
Keywords: breast cancer (BC), DNA methylation, GlaI-PCR assay
DOI: 10.26213/SE.2019.66.12.001
Citation::
Akishev A.G., Netesova N.A., Abdurashitov M.A., Vihlyanov I.V., Nikitin M.K., Karpov A.B., Degtyarev S.Kh. (2019) GlaI-PCR analysis of methylation of ACGC site in Chr11: 65647266 in DNA samples from the blood cells of healthy donors and early stage breast cancers. Epigenet DNA diagnx, vol. 2019(1), DOI: 10.26213/SE.2019.66.12.001

This work is available at Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International
Introduction
Breast cancer (BC) is the most widespread oncological disease among women. According to statistics 10% of women face this disease. 1.38 millions of new breast cancer diagnoses and 460 000 cases of death because of BC are registered by WHO annually in the world [1,2]. One of the main causes of BC high mortality is a late detection of the disease. This is due to the fact that existing methods of BC diagnosing (palpation, mammography, tomography, ultrasound, etc.) are not able to detect small tumors, often located deep in the breast tissue. In this regard, there is an urgent need to develop methods for early detection of breast cancer, allowing to determine the presence of the disease at an early stage.
Currently, one of the most promising areas of cancer diagnostics is epigenetic DNA diagnostics, in particular, determining the methylation status of certain cytosine bases in the regulatory elements of genes in blood DNA preparations. When a tumor is formed, the methylation status of these cytosines is changed; this change can be either demethylation of 5-methylcytosine or methylation of cytosine. The process of changing the methylation status occurs at the early stages of the disease [3,4] and makes this feature particularly valuable in terms of diagnostic potential.
De novo DNA methylation, including aberrant methylation, is carried out by Dnmt3a and Dnmt3b DNA methyltransferases, which recognize site RCGY and modify the internal CG dinucleotide to form the R(5mC)GY sequence in both DNA strands [5], where R is A or G, Y – T or C, 5mC – 5 – methylcytosine. The methylation status of these sites is maintained after the DNA replication by Dnmt1 DNA methyltransferase. The methyl-dependent site-specific DNA endonuclease GlaI recognizes namely R(5mC) ¯ GY site and cleaves it as indicated by the arrow [6], which makes it a convenient tool for identifying de novo methylated sites in human DNA.
Based on the GlaI enzyme, we developed a method of real time GlaI-PCR assay that allows to detect the presence of DNA molecules with unmethylated RCGY site located inside the human genome fragment being studied [7]. GlaI-PCR analysis is performed in two stages and includes a) DNA hydrolysis by the GlaI enzyme and b) real-time PCR from genomic primers bordering the analyzed RCGY site. The use of a fluorescently labeled probe that is complementary to the DNA region between genomic primers significantly increases the specificity of real-time PCR.
GLAD-PCR analysis showed that in more than 82% of donors, the level of ACGC methylation in the SIPA1 gene is 3% or less, while in approximately 70% of patients with breast cancer, the level of ACGC site methylation in SIPA1 gene is in the range of 3.2 to 6.4%. These data suggest that at early stages of breast cancer, demethylation of A(5mC)GC site in the SIPA1 gene at position 65647266 takes place in 2/3 of patients.
The obtained data may be used to develop a PCR test system that allows to exclude diagnosis “breast cancer at early stages” and includes: a) real-time PCR to determine the molar concentration of the DNA preparation [7] from the light fraction of blood cells [8], b) GlaI-PCR assays to determine the concentration of the unmethylated ACGC site in the 11th chromosome at position 65647266, c) determination of proportion of the unmethylated site in the analyzed DNA preparation.
Materials and Methods
Materials
We used bidistilled water, normal saline for injection (JSC “Krasfarma”, Krasnoyarsk), phenol (JSC “Reactiv”, Novosibirsk), chloroform (JSC “Reactiv”, Novosibirsk), isoamyl alcohol (JSC “Reactiv”, Novosibirsk), RNAse A (OOO “Samson-Med”, St. Petersburg), SDS (“Helicon”, Moscow). GLAD-PCR assay Kit, a kit for DNA purification from a light fraction of blood cells, proteinase K, λ DNA, DNA of Raji and U-937 cell lines, restriction endonuclease MspI, Taq I, VspI and buffers – produced by SibEnzyme Ltd. (Novosibirsk, Russia). In the control experiments, genomic DNA from the liver of male A / He mice aged 5-6 months was used. The mouse DNA was isolated as described previously [9]. VspI hydrolysate of DNA from Raji human cell line was obtained by digestion of 10 µg of Raji DNA with VspI in 300 µl of SE-Buffer W at 37ºC for 1 hour followed by phenol-chloroform purification as described previously [9]. DNA samples isolated from a light fraction of blood cells of 19 patients with breast cancer and 28 conditionally healthy donors were studied. Preparation of a light fraction of blood cells and DNA purification were performed as described in the manufacturer’s Protocol [8] with some modifications
Methods
Peripheral blood sampling was performed from the cubital vein in the morning on an empty stomach. When collecting blood, Vacuette, 38 x 0.8 mm, 21Gx11 / 2 needles and 9 ml Vacuette tubes with EDTA (EDTA K2 or EDTA K3) were used. Blood vacuuming unit (immediately after collection) was carefully turned 7-8 times and centrifuged for 3 minutes at 1000 g in an ELMI CM-6MT centrifuge (Latvia). Plasma was carefully collected from each Vacuette tube by pipette with a cut off filter tip into a 15 ml graduated tube, trying to avoid stirring up the blood cell sediment and red blood cell trapping. The tubes were balanced in pairs with saline and centrifuged for 10 minutes at 1000g (ELMI). The supernatant was collected by pipette and discarded. 3 ml of normal saline was added to the each tube. The precipitate was carefully suspended by pipette with a cut off tip. Then 10 ml of normal saline was added to the each tube, gently mixed by inverting and centrifuged for 10 minutes at 1000g (ELMI). The supernatant was collected by pipette and discarded. The resulting cell pellet was stored at -20°C. DNA was isolated from precipitate as described below, either immediately after its preparation or after storage at -20°C.
DNA isolation from the precipitate was carried out with the K008S (SibEnzyme) kit for isolation of DNA from a light fraction of the blood cells according to the manufacturer’s protocol. 1.6 ml of solution No. 1 was added to the precipitate blood cells in the test tube. The sediment was suspended by pipetting to small fragments within 1-2 minutes. The obtained suspension (~ 1.7 ml) was divided equally and the halves were placed in two 2 ml tubes, the tubes were incubated for 30 minutes in a thermostat at 55 ºС. During this time the tubes were occasionally mixed by inverting to completely dissolve the fine particles. Then 300 μl of solution No. 2 were added to each tube, the tubes were gently mixed by turning 2-3 times, placed in a freezer at -20 ° C for 7 minutes and centrifuged for 7 minutes at 13,000 rpm (Eppendorf). The supernatant from the each tube (~ 1 ml) was collected in a new 2 ml tube, 900 μl of solution No. 3 was added to the tube, the mixture was mixed by turning the tubes 3 times. Then the tubes were centrifuged for 5 minutes at 13,000 rpm (Eppendorf). The supernatant was carefully collected by pipette and removed.
The precipitate was washed out in 500 µl of 70% ethanol by turning the tubes 3-4 times and was centrifuged for 1 minute at 13,000 rpm (Eppendorf). The supernatant was carefully collected by pipette (without leaving a liquid in a tube) and removed. The DNA precipitate washing out with 70% ethanol was repeated one more time. Then the precipitate was dried – open tubes were put on the desktop for 10-15 minutes.
The DNA precipitates in 2 tubes (from the same blood cells sample) were dissolved in a mixture of 450 μl of water and 10 μl of TE buffer. 55 µl of 10xSE-buffer Y, 2 µl of Taq I restriction enzyme and 3 µl of RNAse A were added to the obtained DNA solution. The tube was mixed and incubated at 65ºС for 40 min.
After incubation the tubes were cooled to room temperature and phenol-chloroform purification was performed as described previously [10]. An equal volume of phenol impregnated with 10 mM Tris-HCl , pH 8.0 was added to the DNA solution. The mixture was mixed intensively and centrifuged for 3 minutes at 12,000 rpm (Eppendorf). The upper aqueous phase was collected into a new 2 ml tube (not capturing interphase) and an equal volume of phenol – chloroform – isoamyl alcohol (ratio 25:24:1) were added to it. The mixture was mixed intensively and centrifuged for 3 minutes at 12,000 rpm (Eppendorf). The upper aqueous phase was collected into a new 2 ml tube and an equal volume of chloroform-isoamyl alcohol (ratio 24:1) were added to it. The mixture was mixed intensively and centrifuged for 3 minutes at 12,000 rpm (Eppendorf). The upper aqueous phase was collected into a new 2 ml tube and extraction with chloroform-isoamyl alcohol mixture was performed one more time. The upper aqueous phase was collected into a new 2 ml tube and 0.1 volume of 3 M potassium-acetate (pH 7.0) and 2.5 volume of 96% ethanol were added to it. The mixture was mixed and left for one hour at -20ºС. In an hour the mixture was centrifuged for 5 minutes at 13,000 rpm (Eppendorf). The supernatant was carefully collected by pipette (without leaving a liquid in a tube) and removed. The precipitate was washed in 300 µl of 70% ethanol (as described above) two times. The supernatant was carefully collected by pipette (without leaving a liquid in a tube) and removed. The sediment was dried on the table 10 – 15 minutes, dissolved in 400 μl of TE buffer and its concentration was determined on a spectrophotometer «NanoVue Plus» (GE Healthcare, Great Britain). In case of a high concentration of the obtained DNA preparations (more than 30 ng/μl), they were diluted with TE buffer to approximately 30 ng/μl.
The obtained pellets of a light fraction of the blood cells of patients with breast cancer were kept at -20 C. Patients with breast cancer were at the clinical stage of the disease as follows: 12 patients had a stage of disease 1a (T1N0M0), 5 patients had a stage 2a (T2N0M0) and 2 patients had a stage 2b (T2N1M0). Informed consent to participate in the study was obtained from all donors of biological material.
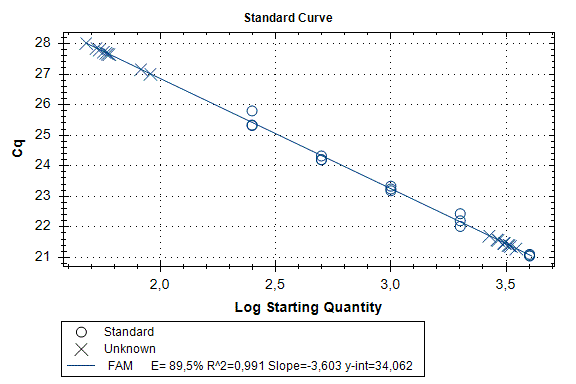
Determination of the concentration of SIPA1 gene fragment and the ACGC site in the 11th chromosome at position 65647266 was performed by real-time PCR in standard 96-well plates with the GlaI-PCR assay kit (“SibEnzyme”). Using this kit, the concentration of molecules containing a fragment of the 11th chromosome at the position 65647215-65647307, as well as the concentration of unmethylated ACGC site in the 11th chromosome at the position 65647266, was determined in each of the studied DNA preparations of the light fraction of blood cells. The study of each DNA sample was carried out in triplets. In case of analysis of N DNA preparations, a reaction mixture was prepared for the analysis of 2N DNA preparations. 1 μl of the obtained DNA preparation and 14 μl of water were added to a tube with the reaction mixture for each triplet. As a result, about 9 ng of DNA (approximately 3000 copies of haploid genome) or less, depending on the concentration of the obtained DNA preparation, was in each PCR well. As a rule a reaction mixture was prepared for the analysis of 12 DNA preparations presented in duplicates (24 test tubes – 72 wells in PCR), mouse DNA (3 wells in PCR) and for construction of a calibration line (5 test tubes – 15 wells in PCR). To prepare the calibration curve (standard curve), VspI hydrolysate of DNA from Raji human cell line was used. The concentration of DNA standard was determined by measuring the optical density at a wavelength 260 nm. Raji DNA standards were prepared as described below. Five calibration tubes and one control tube were prepared as follows:
26 μl of water was added to tube 1;
15 μl of water was added to tubes 2, 3, 4, 5.
14 μl of water was added to tube 6.
4 μl of VspI hydrolysate of Raji DNA at a concentration 18 ng/μl was added to tube 1, mixed by pipetting up and down, with following quick spin-down in a microcenrifuge.
15 μl of the DNA solution from tube 1 were transferred to tube 2 and gently mixed.
15 μl of the DNA solution from tube 2 were transferred to tube 3 and gently mixed.
15 μl of the DNA solution from tube 3 were transferred to tube 4 and gently mixed.
15 μl of the DNA solution from tube 4 were transferred to tube 5 and gently mixed.
15 μl of the DNA solution from tube 5 were removed.
1 μl of mouse liver DNA solution at a concentration of 18 ng/μl was added to tube 6 and gently mixed. All the tubes were centrifuged to get rid of the drops.
A solution A was prepared as follows (calculation is provided per one triplet): 4,2 μl of water, 2,3 μl of 10xTMN buffer, 0,03 μl of lambda DNA (18 ng/μl), 0,2 μl of BSA (10 mg/ml), 0,1 μl of β-mercapthethanol at a concentration 0,2 M and 0,12 μl MspI restriction enzyme (20 units/ μl) . The solution A was mixed, then 6,6 μl of the solution A was added to each tube with DNA sample and to the each calibration tubes. The tubes were incubated 30 minutes at 370C followed by heating 15 minutes at 650C and quick spin-down in a microcenrifuge.
The reaction mixture B was prepared from components of the GlaI-PCR analysis kit and contained (based on 1 triplet) 8.0 µl of water, 0.85 µl of 10xTMN buffer, 0.01 µl of lambda phage DNA (18ng/µl), 0.09 µl of BSA (10 mg/ml), 0.05 µl of β-mercaptoethanol (0,2M) and 0.08 µl of MD DNA endonuclease (20ed/µl).
After incubation and heating at 650С (see above) 8.6 µl of reaction mixture B were added to each tube and incubated for 40 min at 37°C. Tubes were shortly spun down in a microcenrifuge. 30.2 µl of reaction mixture containing (per 1 triplet) 20.8 µl of water, 6.3 µl of 10xGLAD buffer, 0.6 µl of BSA (10 mg/ml), 1.2 µl of dNTP (10 mM each), 2.4 µl of mixture of primers and probe (10 µM each) and 0.4 µl of SP-Taq DNA polymerase (5 u/µl) was added to each tube. After mixing, the contents of each tube (60 µl) were dispensed into the wells of the PCR plate (20 µl in each). PCR plate were sealed, centrifuged and placed in PCR-machine “CFX-96” (Bio-Rad Laboratories, USA).
Nucleotide sequences from the GenBank database (http://ncbi.nlm.nih.gov/genbank) according to the version of the human genome GRCh38/hg38, the set of programs “Vector NTI 11.5” (Invitrogen, USA) and the online resource “BLAST” (http://blast.ncbi.nlm.nih.gov) were used to design a structure of specific primers and probes. A structure of the primers and fluorescently labeled probe used in the work is shown below:
SIPA1F 5′ TGG TGC CCT CGG GTA AGC G 3′
SIPA1R 5′ CCC AGG CCA CAC GGA ACT TTC 3′
SIPA1Z 5′ FAM-CCA CCG CCC GAG GGA TGA AGA CC-BHQ1 3’
Real-time PCR of Raji DNA standards and duplicates of 12 DNA samples of light fraction of blood cells was performed as follows: at 95°C – 3 min; 5 “blind” cycles: 95°C — 10 sec, 67°C (with a decrease of 0.5°C in each subsequent cycle — 20 sec, 72°C — 5 sec, 75°C — 5 sec; 40 cycles: 95°C — 10 sec, 64°C — 20 sec (with detection of a fluorescent signal in the FAM channel), 72°C — 5 sec, 75°C — 5 sec.
After PCR, a number of DNA molecules with a fragment of SIPA1 gene and a number of DNA molecules with unmethylated ACGC sites located in the 11th chromosome at the position 65647266, in 0.33 µl of the studied DNA preparations were determined using the software “Bio-Rad CFX Manager V. 2.1”.
Results and Discussion
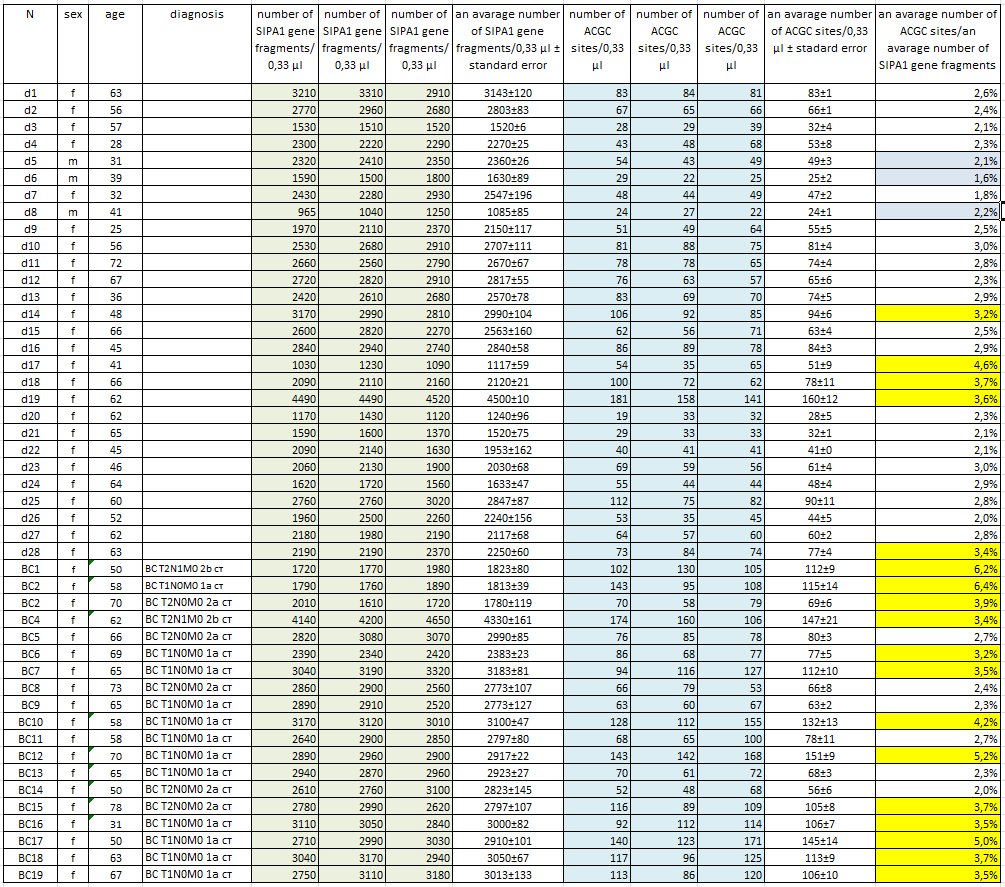
Isolation of DNA from a light fraction of the blood cells of donors and BC patients. Preparation of a light fraction of the blood cells of patients and donors and subsequent DNA isolation was carried out as described in “Materials and Methods”. Blood sampling was performed in the morning on an empty stomach from a vein to vacutainers with EDTA. DNA was purified using a kit for DNA isolation from a light fraction of the blood cells according to the manufacture instructions with some changes as described in “Materials and methods”. The procedure of the cells pellet preparation and DNA purification takes 5-6 hours. In our work, DNA was isolated from a light fraction of the blood cells of 19 patients with BC and 28 conditionally healthy donors. Blood sampling was carried out only from the primary patients at their admission to the hospital. Histological data on the tumor (TMN) were obtained after surgery. Table 1 presents data on donors and patients including the number (column 1), sex (column 2), and age (column 3). Column 4 shows TMN data of patients with breast cancer. The donors included 25 women and 3 men between the ages of 28 and 72. In the list of patients with BC an age of women ranged from 31 to 78 years. At the same time, as can be seen from the table, 17 from 19 DNA preparations were obtained from early stage breast cancer patients (up to stage 2B).
Table 1
Data on DNA preparations and results of GlaI-PCR assay of SIPA1 gene fragment and ACGC site within it.
Structure of the studied fragment of the SIPA1 gene. The DNA fragment studied in this work is located in the 11th chromosome in the region of the SIPA1 gene at the position 65647215-65647307. The structure of the analyzed fragment is shown in figure 1, where the positions of the primers SIPA1F (yellow) and SIPA1R (brown), the SIPA1z probe (green) and the analyzed ACGC site (red) are indicated.
Figure 1. Structure of the analyzed fragment of SIPA1 gene

Determination of the number of the SIPA1 gene fragment copies in the DNA preparations of the light fraction of blood cells. Figure 2 shows the curves of fluorescence accumulation during real-time PCR in course of determination of the number of the SIPA1 gene fragment copies in VspI hydrolysates of Raji DNA at concentrations of 4000, 2000, 1000, 500, and 250 copies/20 µl, which were used as standards in this experiment. The figure also shows the fluorescence accumulation curves for DNA preparations from Raji and U-937 cell lines, as well as the D25 donor at a concentration of 3000 copies/20 µl. Each DNA sample was analyzed twice: with cleavage at site A(5mC)GC and without hydrolysis of this site. As described in “Materials and methods”, the reactions were performed in triplets and a second part of the figure shows the obtained Cq values for each point in the triplet. By comparison with the Cq values obtained for the standards, the number of copies of the SIPA1 gene fragment in each experiment was determined, as well as the average value of the number of copies in the triplet. As can be seen from the presented results, the number of copies of the SIPA1 gene fragment in the analyzed preparations of donor DNA and cell lines is about 3000 in 20 µl, however, this value significantly decreases after cleavage of A(5mC)GC site and only 1.7% – 2.8% of DNA fragments remains intact.
Figure 2. Results of GlaI-PCR assay of d25 donor DNA, Raji DNA and U-937 DNA.
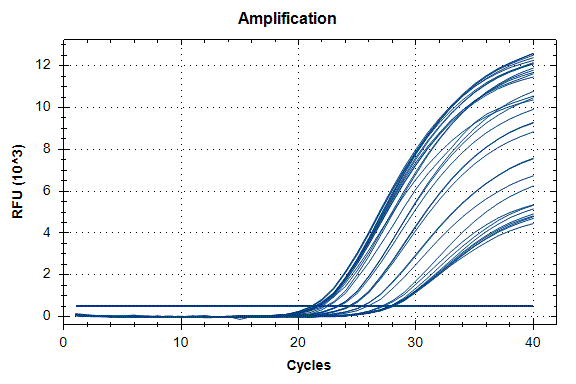
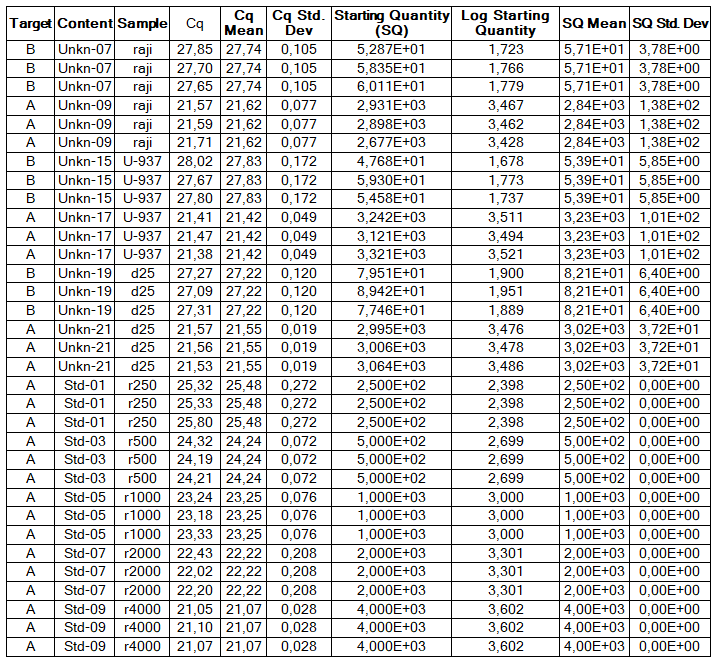
here:
A – SIPA1 DNA fragment in Chr11: 65647215 – 65647307
B – SIPA1 DNA fragment in Chr11: 65647215 – 65647307 cleaved at A(5mC)GC in position 65647266
Thus, concentration of analyzed preparations of donor and cell lines DNA is approximately the same, and the ACGC site in the SIPA1 gene is methylated by more than 97%.
At the next stage the number of copies of the SIPA1 gene fragment and the number of ACGC sites in 0.33 µl of the studied DNA preparations were determined in three experiments, for each of them the reaction mixture was prepared separately. At the same time, as described in “Materials and methods”, in each experiment, the reaction was carried out in a triplet and the average value of the parameter was calculated.
Table 1 shows the determined values of the number of copies of the SIPA1 gene fragment (columns 5, 6, 7) and the number of ACGC sites (columns 9, 10, 11) in 0.33 µl of each DNA sample obtained by real-time PCR using the GlaI-PCR assay kit.
Column 8 and 12 show the average concentrations of the SIPA1 gene fragment and the ACGC site, respectively, for each DNA sample with indication of the standard error. As can be seen from Table 1, the obtained average values of the concentration of the SIPA1 DNA fragment (and, accordingly, genomic DNA) vary from 1085 (d8) to 4500 (d19) copies in 0.33 µl. At the same time, the number of DNA molecules containing unmethylated site ACGC in the 11th chromosome at position 65647266 varies from 24 (d8) to 160 (d19) in 0.33 µl.
Determination of the proportion of DNA molecules containing unmethylated site ACGC in chromosome 11 at position 65647266. The number of DNA molecules containing unmethylated site ACGC in chromosome 11 at position 65647266 was determined in relation to the total number of DNA molecules in the studied samples. This value was calculated dividing the average concentration of unmethylated ACGC sites (column 12 in table 1) by the average number of copies of genomic DNA in 0.33 µl (column 8 in table 1). In Table 1, column 13 shows the obtained values for the percentage of DNA molecules containing the unmethylated ACGC site on chromosome 11 at position 65647266.
As can be seen from the Table, less than 7% of the unmethylated ACGC sites in the SIPA1 gene are detected in all DNA samples. Thus, in the studied DNA samples, more than 93% of the sites in the SIPA1 gene are in the methylated form A(5mC)GC. The data in the Table also shows that in the most donor DNA preparations, the percentage of unmethylated sites is even lower (3% or less), which corresponds to the data presented in Figure 2. The percentage of unmethylated site ACGC in the SIPA1 gene in three male donors (d5, d6, and d8) does not significantly differ from this value in women and ranges from 1.6 to 2.2.
Meanwhile, 5 of the 28 donor DNA samples (less than 18%) have a methylation level of 3.2 to 4.6%. At the same time, only 6 of the 19 DNA samples of patients with breast cancer have a percentage of the unmethylated ACGC site in SIPA1 gene at the level of 2.0% – 2.7%, while the remaining 13 DNA samples of patients with breast cancer (about 70%) have a percentage of unmethylated sites ranging from 3.2% to 6.4%. The data obtained allow to suggest that in case of approximately 2/3 of patients with early stages breast cancer, DNA of blood cells have demethylation of the A(5mC)GC site in SIPA1 gene at position 65647266.
The ACGC site in chromosome 11 at position 65647266 is located in the intron of the SIPA1 (signal-induced proliferation-associated 1) gene. According to the GeneHancer database [11] this position is located in the area of the GH11J065646 enhancer, which includes a binding site of the transcription factor CTCF. It was previously shown that methylation of the CTCF binding site leads to blocking of the enhancer function of this DNA site [12]. Apparently, in case of two-thirds of patients with breast cancer at the initial stages, in the DNA of blood cells involved in the process of recognition and destruction of the tumor cells, there is a demethylation of the site A(5mC)GC in the area of the enhancer GH11J065646, which leads to its activation [13].
The data presented in this paper can be used to develop a PCR test system that allows excluding breast cancer at early stages. All the work to analyze and exclude the diagnosis of breast cancer consists of the following steps:
- preparation of the light fraction of blood cells,
- isolation of DNA from the light fraction of blood cells,
- real-time PCR determination of genomic DNA concentration,
- determination by GlaI-PCR analysis of the concentration of the unmethylated site ACGC at position Chr11: 65647266 in DNA preparations from the light fraction of blood cells and
- calculation of the proportion of DNA molecules containing an unmethylated ACGC site, expressed as a percentage of the total number of DNA molecules.
The GlaI-PCR assay does not require complex and expensive equipment and reagents, the study may be performed in a standard PCR laboratory during annual health examinations and blood tests.
We believe there is a change in methylation of other RCGY sites in the DNA of blood cells, which are involved in immune processes in course of breast carcinogenesis. A use of such sites (along with a studied ACGC at position Chr11: 65647266) for GlaI-PCR assay allows to develop a PCR test system with higher sensitivity and specificity. It is also interesting to study the methylation of RCGY sites and, in particular, ACGC site in the SIPA1 gene, in case of other diseases. The use of GlaI PCR assay to determine the changes in the level of methylation of RCGY sites in DNA preparations from a light fraction of blood cells may allow to develop the diagnostic method for excluding diseases, similar to the one described in this paper.
Patent is pending
References
- World Cancer Report 2014 / B.W. Stewart, C.P. Wild (Eds.). Lyon, International Agency for Research on Cancer.
- Torre L.A., Bray F., Siegel R.L. et al. Global cancer statistics, 2012 // CA Cancer J. Clin. – Vol. 65. – P. 87-108.
- Kisseljova NP, Kisseljov FL. DNA demethylation and carcinogenesis. Biochemistry (Mosc). 2005 Jul;70(7):743-52. Review
- de Caseres I.I., Cairus P. Methylated DNA sequences for early cancer detection, molecular classification and chemotherapy response prediction // Clin. Transl. Oncol. – – Vol. 9. – P. 429-437.
- Handa V., Jeltsch A. Profound flanking sequence preference of Dnmt3a and Dnmt3b mammalian DNA methyltransferases shape the human epigenome // J. Mol. Biol. – – Vol. 348. – P. 1103-1112.
- Tarasova G.V., Nayakshina T.N., Degtyarev S.Kh. Substrate specificity of new methyl-directed DNA endonuclease GlaI // BMC Mol. Biol. – – Vol. 9:7.
- EV Dubinin, AG Akishev, MA Abdurashitov, SB Oleynikova, VL Sitko, and S Kh Degtyarev Real time GlaI-PCR assay of regulation regions of human genes HDAC4, RARB and URB1 // Research Journal of Pharmaceutical, Biological and Chemical Sciences, vol 7(2), p.p. 667-676 (2016).
- Blood light fraction DNA extraction kit // Accessed 28.11.2019 http://russia.sibenzyme.com/info1561.php
- Chernukhin V.A, Abdurashitov M.A., Tomilov V.N., Gonchar D.A., Degtyarev S.Kh. Comparative analysis of mouse chromosomal DNA digestion with restriction endonucleases in vitro and in silico// Ovchinnikov bulletin of biotechnology and physical and chemical biology V.3, No 4, p. 19-27.(2007) (In Russian)
- GlaI PCR assay kit instruction manual // Accessed 28.12.2019 http://sibenzyme.com/info7936.php
- Fishilevich S, Nudel R, Rappaport N, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017;2017:bax028
- Bell AC, Felsenfeld G (May 2000). “Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene”. Nature. 405 (6785): 482–5
- Kim S, Yu NK, Kaang BK. CTCF as a multifunctional protein in genome regulation and gene expression. Exp Mol Med. 2015;47(6):e166.
